Struttura della Materia
lezioni 49 e 50
- riepilogo dei simboli usati per identificare le configurazioni elettroniche ("termini") di una molecola biatomica (qui una buona discussione delle simmetrie per chi vuole approfondire); quasi tutte (non tutte) le molecole biatomiche hanno stato fondamentale con Λ = 0, ma tutte hanno qualche stato eccitato con Λ ≠ 0
- quando c'è un solo elettrone (come in H2+) lo stato σ (λ = 0) non ha un piano nodale che contiene l'asse molecolare; con due o piú elettroni, invece, si può ottenere uno stato Σ (ovvero Λ = 0) anche a partire da stati di singola particella con λ ≠ 0, per esempio riempiendo con due elettroni di opposta proiezione di momento angolare uno stato legante π u; alla presenza o meno di questo piano nodale si riferisce l'apice ± sul simbolo degli stati Σ
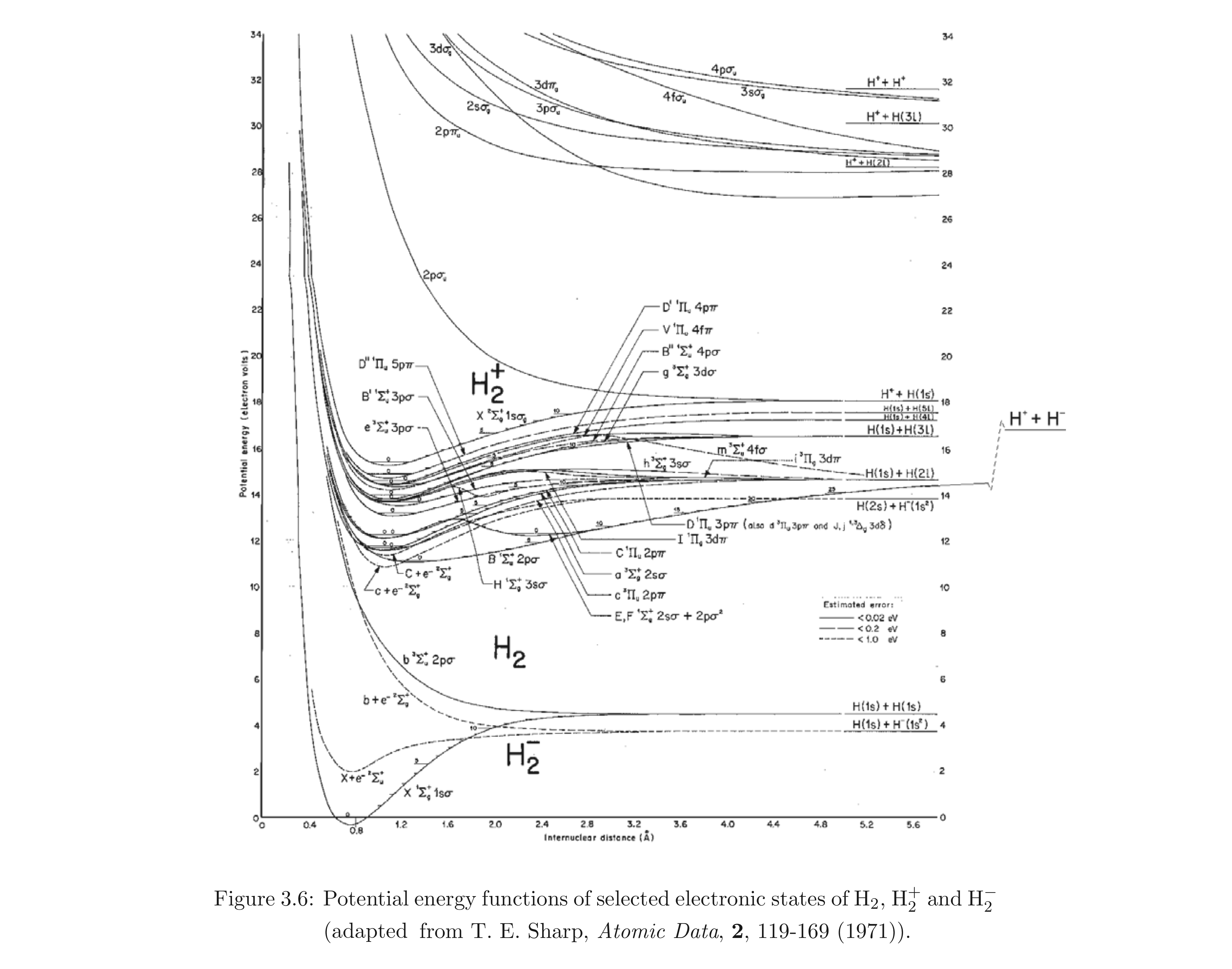
- esempio: fra gli stati elettronici di Born e Oppenheimer dei tre ioni della molecola piú semplice di tutte (H2+, H2 e H2‐), insieme molto piú ampio di quelli da noi visti finora (vedi figura) ce ne sono diversi con Λ ≠ 0
- ultima puntata sulle regole di selezione delle molecole biatomiche, con colpo di scena: come si legge su tutti i testi ma subito si dimentica, l'energia elettronica ε, quando Λ ≠ 0, non dipende piú soltanto dal modulo della distanza internucleare, ma dal vettore; l'equazione di Born-Oppenheimer per la coordinata internucleare non si separa piú in parte radiale e parte angolare, ℓR non è piú un buon numero quantico e non rimane in piedi quasi nessuna delle regole di selezione viste finora (sia per le transizioni roto-vibrazionali, sia per le transizioni elettroniche)
- per determinare le regole di selezione in approssimazione di dipolo nel caso Λ ≠ 0 occorre quindi ripartire da capo (sono conservati il momento angolare totale e la proiezione del momento angolare elettronico sull'asse molecolare ma non il momento angolare nucleare da solo); a questo punto chi vuol approfondire, oltre alla rassegna di van Vleck già segnalata, trova in questo capitolo una discussione abbastanza soddisfacente; gli altri possono accontentarsi di sapere che per le transizioni roto-vibrazionali anche la branca Q risulta permessa quando lo stato fondamentale ha Λ ≠ 0 (ma salvo il radicale OH e pochi altri casi le molecole biatomiche hanno stato fondamentale Σ) e, per le transizioni elettroniche, riferirsi a questa tabella, a posteriori comprensibile alla luce della conservazione del momento angolare totale del sistema fotone+molecola (vedi ultima pagina della presentazione di qualche lezione fa)
- in risposta alla domanda di una studentessa: cenni alla diffusione (scattering) di Rayleigh e alla diffusione (scattering) di Raman e relative regole di selezione (ΔnR = ΔℓR = 0 per Rayleigh ; ΔnR = ±1, ΔℓR = 0, ±2 per Raman); qui
un testo dove chi vuole può approfondire
- il trimero H3++, seconda parte (testo EFAMS pagine 111-112)
- modello di Hubbard della molecola di idrogeno neutra, seconda parte: Hartree-Fock e Heitler-London come limiti opposti nel rapporto fra U, repulsione coulombiana media fra due elettroni che occupano lo stesso orbitale atomico, e t, elemento di matrice off-diagonale dell'hamiltoniana a un elettrone che misura la propensione a "saltare" da un sito atomico all'altro (Ashcroft-Mermin pagina 691)
- svolgimento in aula del compito di esonero di fisica molecolare del 25 maggio 2016, domande 1.1 ‐ 1.4 (qui testo e soluzioni)
{kind=link}