Correzioni anarmoniche e centrifughe: potenziale di Morse
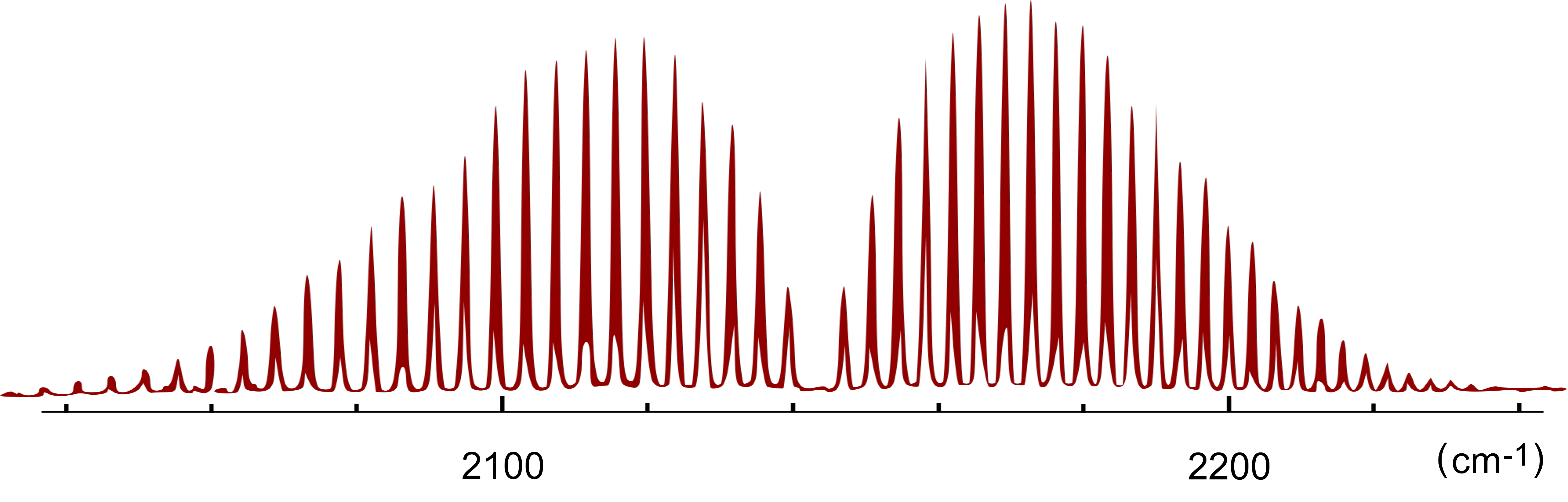
- l'approssimazione armonica e l'approssimazione di rotatore rigido funzionano bene per quasi tutte le molecole biatomiche, ma negli spettri rotovibrazionali sperimentali (per esempio in quello di CO) si apprezza anche a occhio nudo qualche piccola deviazione rispetto a quanto previsto da queste due approssimazioni, specie quando gli stati coinvolti nelle transizioni sono lontani dallo stato fondamentale
- le deviazioni sono dovute al fatto che il rotatore non è davvero rigido (la distanza fra i nuclei oscilla nel tempo) e che il vero potenziale internucleare di Born e Oppenheimer è armonico solo vicino al minimo: al crescere della distanza cresce meno del potenziale armonico (anziché crescere indefinitamente tende a una costante, l'energia dei due atomi isolati) mentre a piccole distanze cresce piú di un potenziale armonico (quando i nuclei si sovrappongono diverge a +∞); come stimare queste deviazioni?
- si sostituisce al potenziale armonico un potenziale modello dotato di comportamento piú realistico; a tale scopo la scelta cade spesso sul potenziale di Morse, uno dei (non molti) potenziali per i quali (per ℓR = 0) esiste una soluzione analitica dell'equazione di Schrödinger; la formula che ne esprime gli autovalori energetici (esatta per ℓR = 0 e ottima approssimazione per per ℓR > 0) è interpretabile in termini di:
- correzioni al rotatore rigido: distorsione centrifuga
- correzioni al modello armonico: anarmonicità (espansione termica)
- correzioni incrociate al rotatore rigido e all'oscillatore armonico
- con opportune definizioni delle costanti questa formula è direttamente utilizzabile per stimare le correzioni anarmoniche e centrifughe di una molecola biatomica reale
NB: le informazioni contenute in questa pagina e nei suoi link sono sufficienti a comprendere il punto e affrontare esercizi; una discussione piú distesa si trova sul Bransden-Joachain
{kind=link}
{kind=link}
{kind=link}